 2018, Vol. 35
2018, Vol. 35文章信息
- 王春龙, 王志刚, 由义敏, 吕智航, 刘泽平, 陈文晶, 史一然
- WANG Chun-long, WANG Zhi-gang, YOU Yi-min, LÜ Zhi-hang, LIU Ze-ping, CHEN Wen-jing, SHI Yi-ran
- 土壤微生物敏感菌及信号调节途径对邻苯二甲酸二甲酯的响应
- Response of the Sensitive Strains and the Signal Regulation Pathway in Soils to Dimethyl Phthalate
- 农业资源与环境学报, 2018, 35(3): 215-221
- Journal of Agricultural Resources and Environment, 2018, 35(3): 215-221
- http://dx.doi.org/10.13254/j.jare.2017.0325
-
文章历史
- 收稿日期: 2017-12-15
录用日期: 2018-01-30

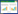




邻苯二甲酸酯(Phthalate acid,PAEs)是一类人工合成的有机化合物,普遍应用于塑料薄膜、农药载体、食品包装、化妆品香味品及润滑剂等多种商品的生产[1-2];同时也应用于聚氯乙烯材料,能使聚氯乙烯由硬塑胶变为弹性的塑胶,在塑料中含量为20%~40%,有的甚至高达55%[3]。PAEs通过氢键和范德华力与塑料基质相结合,因此极易与塑料分离进入环境中,对土壤、水等环境造成危害,植物体可富集并积累PAEs,通过食物链危害动物和人类[4]。邻苯二甲酸二甲酯(Dimethyl phthalate,DMP)是一种被广泛应用的PAEs类物质,广泛存在于大气、水、土壤环境中[5-7],研究发现,广州、深圳地区菜地土壤中PAEs总含量为3~45. 67 mg·kg-1,其中DMP含量远超美国土壤PAEs污染控制标准[8]。土壤中DMP的存在会导致土壤呼吸及相关酶活性发生改变,进而影响土壤健康[3]。因此,中国环境监测中心和美国环保署早已将DMP列为优先控制污染物之一[9]。
土壤微生物是土壤营养物质循环和能量流动中关键的组成部分和主要的驱动力,其与土壤肥力及土壤健康程度都有密切关系[10],还可以调节土壤有机质的分解、腐殖质的形成和营养元素的循环等[11]。与土壤物理化学性质相比,土壤微生物对土壤环境变化的响应更明显,可更快速和精准地对土壤环境条件的变化作出反应,较早地预测土壤环境污染状况的变化过程[12]。本试验研究土壤微生物敏感菌种的丰度及基因调节途径对DMP污染的响应变化,从而进一步阐释DMP对土壤微生物的环境行为,为了解DMP对土壤微生物的生态毒理效应及生物修复技术提供理论基础。
1 材料与方法 1.1 试验土壤制备DMP(纯度>99.9%)购于天津市光富精细化工研究所,丙酮(纯度>99.5%)助溶,制成5 g·L-1的DMP储备液,于4 ℃的冰箱内避光保存备用。取20 kg过0.30 mm筛的土壤,在25 ℃的黑暗培养箱中预培养7 d,恢复土壤微生物活性。试验土壤的基本性质如表 1所示。在1 kg土壤中,加入200 mL DMP储备液,使各样品土壤的DMP质量分数达到1 000 mg·kg-1。待丙酮挥发后,分别取0、0.05、0.1、0.2、0.4 kg的上述土壤和1、0.95、0.9、0.8、0.6 kg的未污染土壤混合均匀,混合后的1 kg样品土壤置于1 L陶瓷花盆中,制成5个浓度的土壤样品,分别为DMP污染对照(CK,0 mg·kg-1)、DMP1(5 mg·kg-1)、DMP2(10 mg·kg-1)、DMP3(20 mg·kg-1)及DMP4(40 mg·kg-1),CK中只加等量的丙酮进行处理,每个处理做3次重复。将土壤样品置于恒温培养箱内培养,用恒重方法保持其土壤的含水量在30%,温度控制在(25±2)℃,培养箱内的相对湿度保持在70%±5%,暗培养20 d之后,取样进行宏基因组测序[5]。
|
取10 g上述培养的土壤样品,使用乙酸乙酯萃取样本,再经旋转蒸发仪提取不同样品的DMP,甲醇洗脱。然后采用液相色谱法测定培养0 d和20 d的土壤样本中DMP的浓度。液相色谱参数:样本量=1 μL;色谱柱为5 μL Eclipse XDB-C18;色谱条件:柱的温度=25 ℃,流动相:甲醇和水,体积比=90:10;探测波长=228 nm。
1.3 DNA提取、测序与数据统计每个样品取10 g土壤使用OMEGA-soil DNA Kit进行土壤微生物DNA提取并用于宏基因组测序和进一步分析[13]。具体方法为:(1)环境样品土壤DNA抽提;(2)通过Covaris M220将所得DNA片段化约为300 bp的短片段;(3)将所得的段片构建Pair End(PE)文库;(4)文库模板进行PCR扩增富集;(5)利用氢氧化钠将富集产物变性,产生单链DNA;(6)利用桥式PCR扩增DNA,产生DNA簇;(7)再将DNA扩增子线性化成为单链;(8)Illumina Hiseq测序。每个样品进行3次上机测序。
统计原始序列,在测序实验中采用了多个样品平行混合测序,各样品中的序列均引入了一段标示其样品来源信息的Index标签序列。根据Index序列区分各个样品的数据,提取出的数据以fastq格式保存。每个样品均有测序两端的Reads,并且Reads的顺序是严格一致、相互对应的。
为了提高后续分析质量和可靠性,对原始测序数据进行清洗处理,将所得数据进行拼接组装,选择最优组装结果。将基因序列聚类,来自相同环境的样品之间有很多微生物(或基因)是共有的,不同基因的丰度在样品之间的变化可以反映样品之间的共性和差异。构建一个非冗余基因集描述该类环境所有基因的整体信息。然后进行基因丰度计算,将每个样品的高质量reads与非冗余基因集进行比对(95% identity),统计基因在对应样品中的丰度信息[13]。
1.4 物种分类学注释所有样品的序列装配为叠连群的数目为4 891~15 857,然后将叠连群预测为ORFs,从CK到DMP4的范围为6 581~24 437。使用BLASTP将基因集与非冗余蛋白质的氨基酸序列数据库(NR数据库)进行比对,并通过NR数据库对应的分类学信息数据库获得物种注释,然后使用物种对应的基因丰度总和计算该物种的丰度,并在域、界、门、纲、目、科、属、种各个分类学水平上统计物种在各个样品中的丰度,从而构建相应分类学水平上的丰度谱[13]。
1.5 微生物群落多样性指数计算宏基因组测序后,得出每个样品的序列(Reads)及操作分类单元(OTUs)的数量。根据所得数据计算香浓(Shannon)指数和辛普森(Simpson)指数[14-15],计算公式如下:
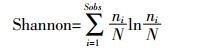
|
(1) |
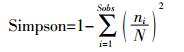
|
(2) |
Sobs为试验得到的OTUs数量;ni为个体的OTUs数量;N为群落中个体总数。
1.6 KEGG功能注释运用BLAST将基因集序列与京都基因和基因组百科全书(KEGG)的基因数据库(GENES)进行比对。根据比对结果使用KOBAS 2.0进行功能注释,并计算对应的基因丰度总和及该功能类别的丰度[13]。
2 结果与分析 2.1 土壤中DMP浓度变化DMP处理土壤后,培养0 d时,测定的DMP浓度与添加的DMP浓度基本一致。培养20 d后,发现DMP浓度显著降低(图 1)。

|
| 图 1 DMP浓度变化 Figure 1 The change of DMP concentration |
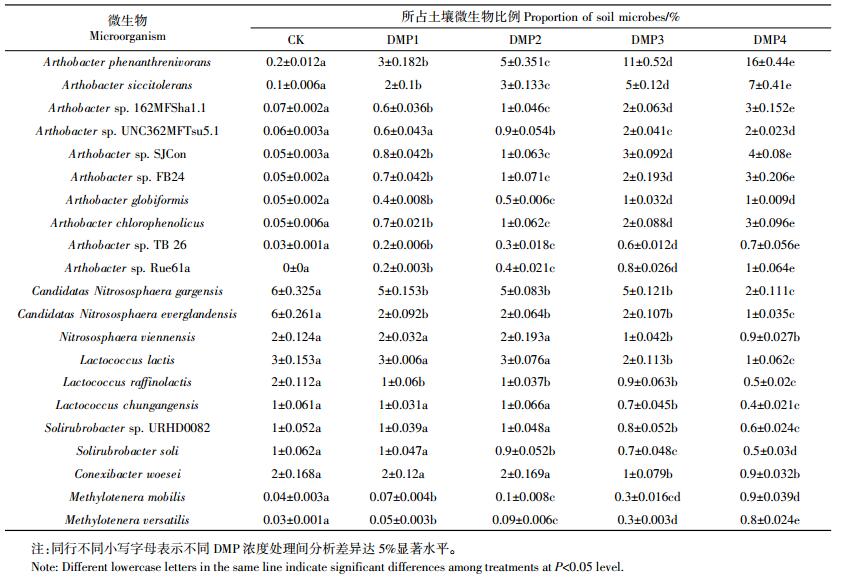
本试验结果发现经DMP污染土壤20 d后,共鉴定出3 855种土壤微生物,将21种显著变化的微生物定义为对DMP敏感菌株,可以发现,随着DMP污染浓度的增加土壤微生物敏感菌种的生长显著增加或抑制(表 2)。其中一些敏感菌种Arthobacter phenanthrenivorans、Arthobacter siccitolerans、Arthoba-cter sp.162MFSha1.1、Arthobacter sp. UNC362MFTsu5.1、Arthobacter sp. SJCon、Arthobacter sp. FB24、Arthobacter globiformis、Arthobacter chlorophenolicus、Arthobacter sp. Rue61a、Arthobacter sp. TB 26、Methylotenera mobilis和Methylotenera versatilis的生长随着DMP污染浓度增加而加快,而有一些敏感菌种的生长受到DMP抑制,主要包括Lactococcus lactis、Lactococcus raffinolactis、Lactococcus chungangensis、Solirubrobacter sp. URHD0082、Solirubrobacter soli、Conexibacter woesei、Candidatas Nitrososphaera gargensis、Candidatas Nitrososphaera everglandensis和Nitrososphaera viennensis。
本试验对通过宏基因组测序得到的结果,进行了土壤微生物群落多样性分析。结果发现Simpson指数和Shannon指数均随DMP污染浓度增加而降低(图 2)。Simpson指数从0.968 0降低至0.764 5,Shannon指数从6.953 8降低至4.749 0。在DMP污染浓度相对较低时(DMP1和DMP2)多样性指数降低比较缓慢,而高浓度DMP污染时(DMP3和DMP4)下降程度较大。

|
| 图中直方柱上方小写字母不同表示处理间差异显著(P < 0.05)。下同 Different lowercase letters indicate significant differences among treatments at P < 0.05 level. The same below 图 2 微生物多样性指数对DMP污染的响应 Figure 2 The response of microbial diversity to DMP contamination |
为了预测土壤微生物的信号调控途径,采用KEGG数据库与所有样品基因组进行比对分析。途径注释结果表明,DMP污染增加了土壤系统的ABC转运系统、双组分系统(TCS)及磷酸转移酶系统(PTS)的总基因丰度,且其丰度随着DMP污染浓度增加而显著升高(图 3A~图 3C)。土壤ABC转运系统的单糖运输ATP酶及MalK和MsmK的基因丰度在DMP污染后显著增加(图 3D和图 3E)。在土壤TCS中,随着DMP污染浓度的增加,苹果酸脱氢酶、柠檬酰-CoA裂解酶和组氨酸激酶的基因丰度也呈增长趋势(图 3F~图 3H)。DMP污染后,土壤PTS中磷酸转移酶及Crr和BglF的基因丰度显著提高,且与DMP浓度呈正相关(图 3I和图 3J)。

|
| 图 3 DMP污染对土壤调节途径的影响 Figure 3 Effect of DMP on regulation pathways in soils |
土壤微生物可以早期预测土壤环境污染的变化过程,参与土壤物质和能量转化等重要过程[16]。宏基因组测序被广泛用于分析原始和污染环境的微生物群落DNA,并评估污染物对原始生态系统的影响[17]。本试验基于宏基因组测序,分析比较CK和DMP处理样品得出各样品的微生物敏感菌种的丰度、调节途径等,从而揭示DMP污染对土壤微生物系统的环境行为。
本文在菌种水平分析土壤微生物群落时筛选出了21株菌种,这些菌种在微生物群落中所占比例较大,且受到DMP污染显著影响,均为敏感菌种。Arthrobacter在土壤中主要功能是对芳香族化合物进行降解,DMP污染土壤后,发现土壤Arthrobacter菌属中Arthrobacter phenanthrenivorans、Arthrobacter siccitolerans和Arthrobacter sp. SJCon菌种在土壤微生物系统中所占的比例显著增加,且与DMP浓度呈正相关[18-19],与DMP降解实验结果一致。Candidatas Nitrososphaera gargensis、Candidatas Nitrososphaera everglandensis和Nitrososphaera viennensis菌种属于Nitrososphaera菌属,这些菌种对土壤氮循环十分重要,而其生长受DMP抑制[20]。Conexibacter woesei菌种在土壤系统中也参与土壤氮循环,对土壤健康极其有益,它们的生长受到DMP抑制[21]。Lactococcus lactis、Lactococcus raffinolactis和Lactococcus chungangensis菌种是丰富的碳氮基质,在各种植物中存在[22]。Solirubrobacter sp. URHD0082和Solirubrobacter soli[23]菌种,可产生活性次生代谢产物,可能对某些菌的生长有促进和抑制作用[24]。Methylotenera mobilis和Methylotenera versatilis属于甲基营养型,是专性甲基应用者并且与有机污染物降解有关[25],且其生长受DMP污染抑制。除与降解有机物有关的菌种外,其余菌种的相对百分比均随DMP浓度的增加而减少。研究结果表明DMP污染抑制了大部分菌种的生长。而且,通过计算Simpson指数和Shannon指数,分析其结果发现DMP污染降低了土壤微生物群落的多样性[15, 26]。因此,DMP污染显著改变了土壤微生物敏感菌种的丰度。本试验结果与Wang等[5]的16S高通量测序(V3~V4区)结果一致。
ABC转运系统存在于所有生物体中,其功能是将底物转运至活细胞,运输的基质包括糖、氨基酸、肽、离子及其他分子,也可促进细胞对营养的吸收[27]。DMP污染增加了ABC转运系统的总基因丰度以及单糖转运ATP酶的MalK和MsmK基因丰度,可能会增加某些菌的代谢及繁殖,也可能加速土壤中营养物质的消耗。在原核生物中,TCS是一种复杂的信号转导通路,它可感知到环境中的任何变化并作出相应的反应[28],可调节下游基因表达,从而协调细胞反应,参与生物化学过程,并可作为能量载体[29]。在DMP污染后,TCS的总基因丰度以及苹果酸脱氢酶、组氨酸激酶以及柠檬酰-CoA连接酶基因的丰度显著增加,表明DMP污染可能导致土壤代谢及能量转换加速。PTS在细菌中是调节营养物质代谢的关键信号转导通路[30],DMP污染增加了PTS的总基因丰度以及磷酸转移酶、Crr和BglF基因的丰度,PTS的变化是菌种丰度改变的原因之一[31]。因此,ABC转运体、TCS和PTS的变化可能引起土壤微生物菌种丰度的改变。
4 结论DMP污染处理土壤20 d后,通过土壤宏基因组测序共发现21株对DMP污染敏感的菌种,其中一部分敏感菌种的生长受到了DMP污染的促进,而另一部分菌种的生长则被抑制;DMP的污染降低了土壤微生物群落的多样性,且随着DMP浓度增大,土壤微生物群落多样性降低;土壤微生物ABC转运系统、TCS和PTS的总基因丰度及相关酶的基因丰度受到DMP污染的促进,且与DMP污染的浓度呈正相关。因此,DMP污染改变了土壤微生物敏感菌种的丰度,进而干扰了土壤系统的生态平衡。
| [1] |
王志刚, 由义敏, 徐伟慧, 等. 黑土微生物丰度和多样性对邻苯二甲酸二丁酯污染的响应[J]. 生态环境学报, 2015, 24(10): 1725-1730. WANG Zhi-gang, YOU Yi-min, XU Wei-hui, et al. Response of the abundance and diversity of microbes in black soil to di-n-butyl phthalate contamination[J]. Ecology and Environmental Sciences, 2015, 24(10): 1725-1730. (in Chinese) |
| [2] |
陶然. 食品中邻苯二甲酸酯类化合物残留分析方法的研究[D]. 芜湖: 安徽工程大学, 2013. TAO Ran. Method for determination of phthalate acid esters in food material[D]. Wuhu: Anhui Engineering University, 2013. (in Chinese) https://wenku.baidu.com/view/1d11f8aef80f76c66137ee06eff9aef8951e4870.html |
| [3] |
王志刚, 胡影, 徐伟慧, 等. 邻苯二甲酸二甲酯污染对黑土土壤呼吸和土壤酶活性的影响[J]. 农业环境科学学报, 2015, 34(7): 1311-1316. WANG Zhi-gang, HU Ying, XU Wei-hui, et al. Impacts of dimethyl phthalate contamination on respiratory rates and enzyme activities in black soil[J]. Journal of Agro-Environment Science, 2015, 34(7): 1311-1316. DOI:10.11654/jaes.2015.07.012 (in Chinese) |
| [4] |
Zeng H H, Zhang H X, Wu X, et al. Pollution levels and health risk assessment of particulate phthalic acid esters in arid urban areas[J]. Atmospheric Pollution Research, 2016, 8(1): 188-195. |
| [5] |
Wang Z G, Hu Y L, Xu W H, et al. Impacts of dimethyl phthalate on the bacterial community and functions in black soils[J]. Frontiers in Microbiology, 2015(6): 405. |
| [6] |
Wang P, Wang S L, Fan C Q. Atmospheric distribution of particulate-and gas-phase phthalic esters(PAEs) in a Metropolitan City, Nanjing, East China[J]. Chemosphere, 2008, 72(10): 1567-1572. |
| [7] |
Zheng X, Zhang B T, Teng Y. Distribution of phthalate acid esters in lakes of Beijing and its relationship with anthropogenic activities[J]. Science of the Total Environment, 2014, 476/477: 107-113. DOI:10.1016/j.scitotenv.2013.12.111 |
| [8] |
蔡全英, 莫测辉, 李云辉, 等. 广州、深圳地区蔬菜生产基地土壤中邻苯二甲酸酯(PAEs)研究[J]. 生态学报, 2005, 25(2): 283-288. CAI Quan-ying, MO Ce-hui, LI Yun-hui, et al. The study of PAEs in soils from typical vegetable fields in areas of Guangzhou and Shenzhen, South China[J]. Acta Ecologica Sinica, 2005, 25(2): 283-288. (in Chinese) |
| [9] |
罗萌, 宋娇艳, 曾微, 等. 邻苯二甲酸二甲酯在紫色土中的淋溶释放特征及其影响因素研究[J]. 地球与环境, 2017, 45(5): 531-539. LUO Meng, SONG Jiao-yan, ZENG Wei, et al. Release leaching characteristics of dimethyl phthalate in purple soil and its influencing factors[J]. Earth and Environment, 2017, 45(5): 531-539. (in Chinese) |
| [10] |
Zhou J, Guan D, Zhou B, et al. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China[J]. Soil Biology & Biochemistry, 2015, 90: 42-51. |
| [11] |
Ding J, Jiang X, Ma M, et al. Effect of 35 years inorganic fertilizer and manure amendment on structure of bacterial and archaeal communities in black soil of northeast China[J]. Applied Soil Ecology, 2016, 105: 187-195. DOI:10.1016/j.apsoil.2016.04.010 |
| [12] |
Hou S, Xin M, Wang L, et al. The effects of erosion on the microbial populations and enzyme activity in black soil of northeastern China[J]. Acta Ecologica Sinica, 2014, 34(6): 295-301. DOI:10.1016/j.chnaes.2014.10.001 |
| [13] |
Ma J, Wang Z, Li H, et al. Metagenomes reveal microbial structures, functional potentials, and biofouling-related genes in a membrane bioreactor[J]. Applied Microbiology & Biotechnology, 2016, 100(11): 5109-5121. |
| [14] |
Schloss P D, Westcott S L, Ryabin T, et al. Introducing mothur:Open-source, platform-independent, community-supported software for describing and comparing microbial communities[J]. Applied & Environmental Microbiology, 2009, 75(23): 7537-7541. |
| [15] |
He F, Hu X S. Hubbell's fundamental biodiversity parameter and the Simpson diversity index[J]. Ecology Letters, 2005, 8(4): 386-390. DOI:10.1111/ele.2005.8.issue-4 |
| [16] |
Tian Y Q, Zhang X Y, Liu J, et al. Microbial properties of rhizosphere soils as affected by rotation, grafting, and soil sterilization in intensive vegetable production systems[J]. Scientia Horticulturae, 2009, 123(2): 139-147. DOI:10.1016/j.scienta.2009.08.010 |
| [17] |
Hemme C L, Tu Q, Zhou S, et al. Comparative metagenomics reveals impact of contaminants on groundwater microbiomes[J]. Frontiers in Microbiology, 2015, 6: 1205. |
| [18] |
Zhang Y, Ge S, Jiang M, et al. Combined bioremediation of atrazine-contaminated soil by Pennisetum and Arthrobacter sp. strain DNS10[J]. Environmental Science & Pollution Research, 2014, 21(9): 6234-6238. |
| [19] |
李娟, UwaremweC, 冷艳, 等. 节杆菌属细菌处理有机物和重金属污染物的研究进展[J]. 环境科学与技术, 2017, 40(10): 89-97. LI Juan, Uwaremwe C, LENG Yan, et al. Progress on the study of biodegradation of organic pollutants and adsorption of heavy metals with Arthrobacter strains[J]. Environmental Science & Technology, 2017, 40(10): 89-97. (in Chinese) |
| [20] |
Zhang J, Liu B, Zhou X, et al. Effects of emergent aquatic plants on abundance and community structure of ammonia-oxidising microorganisms[J]. Ecological Engineering, 2015, 81: 504-513. DOI:10.1016/j.ecoleng.2015.04.029 |
| [21] |
Anderson C R, Hamonts K, Clough T J, et al. Biochar does not affect soil N-transformations or microbial community structure under ruminant urine patches but does alter relative proportions of nitrogen cycling bacteria[J]. Agriculture Ecosystems & Environment, 2014, 191(1): 63-72. |
| [22] |
Sanders J W, Venema G, Kok J. Environmental stress responses in Lactococcus lactis[J]. FEMS Microbiology Reviews, 1999, 23(4): 483-501. DOI:10.1111/j.1574-6976.1999.tb00409.x |
| [23] |
Zhang T, Liu M, Sun J, et al. Bacterial diversity in rock varnish of extreme arid region of Turpan Basin[J]. Acta Ecologica Sinica, 2012, 32(5): 265-270. DOI:10.1016/j.chnaes.2012.07.006 |
| [24] |
Ettoumi B, Chouchane H, Guesmi A, et al. Diversity, ecological distribution and biotechnological potential of Actinobacteria inhabiting seamounts and non-seamounts in the Tyrrhenian Sea[J]. Microbiological Research, 2016, 186/187: 71-80. DOI:10.1016/j.micres.2016.03.006 |
| [25] |
Jiao S, Liu Z, Lin Y, et al. Bacterial communities in oil contaminated soils:Biogeography and co-occurrence patterns[J]. Soil Biology & Biochemistry, 2016, 98: 64-73. |
| [26] |
Jurasinski G, Retzer V, Beierkuhnlein C. Inventory, differentiation, and proportional diversity:A consistent terminology for quantifying species diversity[J]. Oecologia, 2009, 159(1): 15-26. DOI:10.1007/s00442-008-1190-z |
| [27] |
Liston S D, Mann E, Whitfield C. Glycolipid substrates for ABC transporters required for the assembly of bacterial cell-envelope and cell-surface glycoconjugates[J]. Biochimica et Biophysica Acta(BBA)-Molecular and Cell Biology of Lipids, 2016, 1862(11): 1394-1403. |
| [28] |
Chang C, Stewart R C. The two-component system. Regulation of diverse signaling pathways in prokaryotes and eukaryotes[J]. Plant Physiology, 1998, 117(3): 723-731. DOI:10.1104/pp.117.3.723 |
| [29] |
Aggarwal S, Somani V K, Gupta V, et al. Functional characterization of PhoPR two component system and its implication in regulating phosphate homeostasis in Bacillus anthracis[J]. Biochimica et Biophysica Acta(BBA)-General Subjects, 2017, 1861(1): 2956-2970. DOI:10.1016/j.bbagen.2016.09.022 |
| [30] |
Gao G, Wang A, Gong B L, et al. A novel metagenome-derived gene cluster from termite hindgut:Encoding phosphotransferase system components and high glucose tolerant glucosidase[J]. Enzyme & Microbial Technology, 2016, 84: 24-31. |
| [31] |
Barabote R D, Jr S M. Comparative genomic analyses of the bacterial phosphotransferase system[J]. Microbiology & Molecular Biology Reviews, 2005, 69(4): 608-634. |