 2014, Vol. 31
2014, Vol. 31文章信息
- 杜东霞, 尹红梅, 张德元, 刘标, 许隽, 王震, 贺月林
- DU Dong-xia, YIN Hong-mei, ZHANG De-yuan, LIU Biao, XU Jun, WANG Zhen, HE Yue-lin
- 发酵床垫料中微生物16S rDNA PCR反应条件的建立与优化
- Establishment and Optimization of Microbial 16S rDNA PCR Reaction Conditions in Fermentation Bed Padding Material
- 农业资源与环境学报, 2014, 31(5): 470-475
- Journal of Agricultural Resources and Environment, 2014, 31(6): 513-520
- http://dx.doi.org/10.13254/j.jare.2014.0102
-
文章历史
- 收稿日期:2014-04-24





随着规模化养猪产业的迅速发展,养殖场造成的 环境污染问题显得日℃突出。而发酵床养殖技术是依 据自然生态理念和微生态原理,集畜禽粪尿无害化处 理与科学饲养于一体的新型环保养猪技术[1]。该养殖 技术是以猪舍内铺设谷壳、米糠、锯末等原料组成的 基质垫料作为培养基,人为接种一些环境℃生菌,猪 饲养过程中所排放的粪尿在猪舍内经微生物原地发 酵迅速降解、消化,从而达到零污染、零排放,从源头 实现环保、无公害的养殖目的[2]。
发酵床养殖技术的精髓是微生物的发酵和代谢 作用,起主要作用的微生物群落结构十分复杂,除了 发酵床垫料中的土著微生物外,还包含人为添加的一 些功能微生物菌群,在饲养环境下,微生物在相互作 用过程中,其群落结构和数量此消彼长,处于不断变 化过程中,最终会产生一些优势菌群,主导着畜禽粪 便的无害化过程[1, 3]。因此,有必要弄清楚成分复杂的 发酵床垫料中微生物的种类、数量、亲缘关系以及群 落的演替规律,有助于开发出新型、高效、安全、稳定 的微生物菌剂,促进发酵床养猪技术的进一步发展。 然而,近几年来,发酵床垫料中的微生物群落动态及 分子生态学的研究显得十分的薄弱,一方面是由于许 多功能微生物无法在实验室培养,传统的微生物纯培养无法反映发酵床垫料中微生物的丰度;另一方面用 分离微生物的方法研究微生物群落动态变化规律工 作十分繁重,而且对了解微生物实际群落变化比较 困难[2]。
近年来,随着现代生物技术和微生物分子生物学 的迅速发展,基于DNA分析的分子生态技术为解决该 难题提供了强有力的工具。通过菌群的DNA分析可以 比较全面地了解发酵床垫料中微生物群落的结构变 化,极大地促进发酵床垫料中微生物多样性的研究。 它既可以绕开传统分离方法复杂和繁琐的操作,又可 以鉴定纯培养方法难以鉴定的菌种[4, 5]。但该方法的应 用必须以获得能真实反映原环境样品中微生物实际 组成状况的总DNA 及高质量的PCR 产物为基础,鉴 于此,本文对发酵床垫料中微生物总DNA 的提取方 法进行了探索,同时建立与优化了微生物16S rDNA PCR 反应条件,为揭示发酵床垫料中微生物群落的动 态变化规律及研制高效专一的发酵床复合菌剂奠定 理论基础。 1 材料与方法 1.1 实验材料 1.1.1 发酵床垫料
来源于唐人神养殖场,在发酵床垫料30 cm 处 “Z”字形5 点取样,混合均匀后作为实验样本,塑料袋 密封低温保存。 1.1.2 主要试剂和设备
土壤总DNA提取试剂盒(UltraCleanSoilDNAIso lation Kit),购于美国MOBIO 公司;PCR 仪(Mastercy cler pro)购于Eppendorf 公司;Cary50 紫外可见分光 光度计购自美国VARIAN 公司;5424R 小型台式高速 冷冻离心机购自德国Eppendorf 公司;DYY-6C 型电 泳仪购自北京六一仪器厂;Bio-Rad Gel Doc 2000 型 凝胶成像系统购自美国Bio-Rad公司。 1.2 实验方法 1.2.1 发酵床垫料中微生物总DNA 提取
参照文献[6]的方法并进行了适当修改。 1.2.1.1 发酵床垫料样品预处理
新鲜垫料经液氮研磨之后,称取5 g于30 mL 离 心管中,用TENP缓冲液洗涤,直到上清液清澈透明, 离心的沉淀用于DNA提取。 1.2.1.2 发酵床垫料中微生物总DNA 提取
采用以下2 种方法对制备的发酵床垫料样品进 行微生物总DNA 的提取。
(1)SDS-CTAB 结合法:①称取2 g 沉淀置于30 mL洁净离心管中,在沉淀中加入15 mL SDS 裂解液, 200 μL 蛋白酶K 和0.8 mL 异硫氰酸胍洗液,轻轻颠 转混匀,65 ℃温育1 h;②6 000 r·min-1离心10 min,转 移上清液至30 mL 洁净离心管;③加入0.2 倍体积的 醋酸钠溶液和1.7 倍体积的10% PEG8000 溶液,-20 ℃下静置15 min;④10 000 r·min-1,4 ℃,离心10 min, 留沉淀;⑤往沉淀中加入15 mL CTAB 裂解液,65 ℃ 温育15 min;⑥加入等体积氯仿颐异戊醇(24:1),轻轻 混匀,10 000 r·min-1离心10 min;⑦将上清液转移到 新的30 mL 离心管中,加入2 倍体积的无水乙醇,置 于4 ℃过夜;⑧12 000 r·min-1,4 ℃,离心10 min,弃上 清液,留沉淀。用1 mL 70%乙醇洗涤沉淀,转移到 1.5 mL离心管中。14 000 r·min-1,4 ℃,离心3 min,弃 上清液,留沉淀。再重复洗涤1 次,超净台吹干,之后 加入300 μL 无菌水溶解DNA,-20 ℃保存备用。
(2)土壤总DNA 提取试剂盒法:该实验步骤严格 参照说明书进行操作。 1.2.2 DNA 浓度和纯度的测定方法
用紫外分光光度法分别测定不同方法提取的 DNA 溶液在不同波长处的吸光度(OD260、OD230、 OD280),根据公式:[dsDNA] = 50×OD260×稀释倍数,计 算DNA的浓度(单位为μg·mL-1),分别用OD260/OD230、 OD260/OD280估算所提取DNA 的纯度。 1.2.3 发酵床垫料中微生物16S rDNA PCR 反应体系的建立
引物:27F(5'-AGAGTTTGATCCTGGCTCAG-3') 和1492R(5'-TACCTTGTTACGACTT-3')扩增细菌 16S rDNA;
PCR 扩增体系:10×PCR buffer 2.5 μL,dNTPs 2 μL,上下游引物(10 μmol·L-1)各1 μL,DNA 模板稀释 液1 μL,Taq DNA 聚合酶(5 U·μL-1)0.2 μL,其余用无 菌水补齐至25 μL;
PCR 反应条件:94 ℃预变性4 min,(94 ℃变性45 s,53℃退火30 s,72 ℃延伸90 s)30个循环,最后72 ℃ 延伸10 min。 1.2.4 发酵床垫料中微生物16S rDNA 的PCR 反应条件优化
退火温度的选择:对扩增程序中最为关键的退火 温度进行优化,分别设置为:48.3、50.0、51.8、53.7、 55.5 ℃。
循环次数的选择:在确定最佳退火温度后,选择 不同循环次数进行扩增。循环次数分别为20、25、30、35、40、45 个。
引物浓度的选择:引物浓度梯度设置为0.2、0.4、 0.6、0.8、1.0 μmol·L-1。
dNTPs 浓度的选择:dNTPs 浓度梯度设置为0.1、 0.2、0.3、0.4、0.5 mmol·L-1。
不同Taq DNA 聚合酶用量的选择:Taq DNA 聚 合酶浓度梯度设置为0.5、0.75、1.0、1.5、2 U。
模板DNA 浓度的选择:模板DNA 浓度梯度设置 为20、30、40、50、60、70 ng。
Mg2+浓度的选择:Mg2+浓度梯度设置为1、1.5、2、 2.5、3.0 mmol·L-1。
PCR 扩增产物用0.8%的琼脂糖凝胶电泳检测。 2 结果与分析 2.1 发酵床垫料中微生物总DNA 的纯度和浓度检测结果
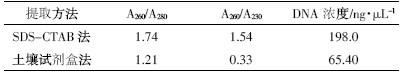
将不同方法处理提取的垫料微生物总DNA 通过 紫外分光光度计测定的数据见表 1。DNA 纯度是衡量 垫料微生物总DNA 提取的重要指标,一般用A260/A280 和A260/A230 的比值分别检测提取DNA 样品中蛋白质 和腐植酸杂质的污染程度[7, 8]。A260/A230比值越大,DNA 越纯;反之A260/A230比值越低,腐植酸污染越严重。一 般来说,当A260/A280 比值大于1.7 时,DNA 较纯,否则 有蛋白质污染[2, 9]。
 |
利用梯度PCR 对最佳退火温度进行筛选,由图 1 可知,随着退火温度的逐渐升高,所对应泳道的目的 条带逐渐明亮、清晰,当退火温度升高至53.7 ℃时条 带达到最亮。但是若温度继续升高至55.5 ℃时几乎 没有目的条带出现。综合考虑,最佳退火温度设置在 53.7 ℃较为合适。

|
| 图 1 不同退火温度对PCR效果的影响 Figure 1 Effectof differentannealing temperatureson PCRproducts |
选择合适的循环次数对扩增结果的影响非常大。 理论上,循环次数越多,扩增产率越高。保持其他程序 设置不变,在退火温度为53.7 ℃时进行20、25、30、 35、40、45 次循环。由图 2 可知,由20 至25 个循环,扩增产物的量逐渐升高,但循环次数过高则扩增效果 并不理想。当循环次数达到30个循环时,扩增的量开 始降低,当再逐渐升高时,几乎看不到单一条带。由此 可知,在该退火温度下的最佳循环次数为25个。

|
| 图 2 不同循环次数对PCR效果的影响 Figure 2 Effect of different cycle times on PCR products |
在最佳退火温度和最佳循环次数下,对最佳引物 浓度进行了筛选,具体引物浓度为0.2、0.4、0.6、0.8、 1.0 μmol·L-1;由图 3可知,在PCR 反应过程中并不是 引物的浓度越高越好,随着引物浓度的增加,PCR 产 物的扩增量并没有增加,当达到0.8 μmol·L-1 时,扩 增量反而降低。所以在PCR 反应体系中,引物浓度过 高不仅造成试剂的浪费,而且会干扰PCR过程。由此 可知,最佳引物浓度为0.2 μmol·L-1。

|
| 图 3 不同引物浓度对PCR 效果的影响 Figure 3 Effect of diferrent primer concentration on PCR products |
已有研究表明,在PCR 扩增中,过高的dNTP 浓 度可能导致碱基错配而出现非特异性扩增,过低浓度又可能会因过早地消耗而使产物单链化或产物量不 足[10]。由图 4 可知,当dNTP 浓度为0.1~0.3 mmol·L-1 时,扩增出的谱带较为一致,其中以0.2 mmol·L-1 浓 度扩增量最多;当dNTP 的浓度等于或高于0.4 mmol·L-1时,看不出单一的条带。由此可知,该反应体 系中dNTP的最佳用量为0.2 mmol·L-1。

|
| 图 4 不同dNTPs浓度对PCR效果的影响 Figure 4 Effect of diferrent dNTPs concentration on PCR products |
一般来说,DNA 模板的适宜用量有一个较大的 范围,在此范围内随着DNA 量的增加条带更加清晰, 但DNA 模板浓度并不是越高越好,DNA 浓度过高反 而增加退火时引物结合的难度,造成PCR 扩增产量 降低。由图 5可知,随着DNA 模板浓度由20~50 ng的 升高,PCR 扩增量也逐渐增多,但当DNA 模板浓度大 于50 ng 时,PCR 扩增条带开始变弱,因此,本反应体 系的最佳DNA 模板浓度为50 ng。

|
| 图 5 不同DNA 模板浓度对PCR 效果的影响 Figure 5 Effect of diferrent DNA template concentration on PCR products |
Taq DNA 聚合酶的种类及其浓度是影响PCR 扩 增的关键因素。过高浓度的Taq 酶不仅会造成不必要 的浪费,且使得非特异性扩增产物的几率增大,过低 的浓度则会降低合成效率。由图 6 可知,0.5~2.0 U TaqDNA 聚合酶的5 个泳道均有扩增产物,而且没有 太大的差异,因此,结合经济角度和实际观测结果,可 确定0.5 U为本实验中Taq DNA 聚合酶的最佳浓度。

|
| 图 6 不同Taq DNA 聚合酶浓度对PCR效果的影响 Figure 6 Effect of diferrent Taq DNA polymerase concentration on PCR products |
影响PCR 反应结果的一个重要因素是MgCl2浓 度,它不仅影响引物与模板的结合效率和引物二聚 体的形成,还影响Taq DNA 聚合酶的活性,Mg2+浓 度对PCR 扩增的特异性和效率影响很大,通常认为 Mg2+的适宜浓度范围在1.5~2.5 mmol·L-1之间[4, 11]。由 图 7 可知,Mg2+浓度在1.0~3.0 mmol·L-1 范围内,除了1 mmol·L-1 Mg2+浓度没有扩增条带之外,其他浓度条 件下均有扩增产物,综合考虑,该反应条件下的最佳 Mg2+浓度为2 mmol·L-1。

|
| 图 7 不同Mg2+浓度对PCR效果的影响 Figure 7 Effect of diferrent Mg2+ concentration on PCR products |
综上所述,以发酵床垫料中微生物总DNA 为模 板,扩增16S rDNA 反应体系中5 个因素的最佳浓度 为:总体积为25μL的反应体系包括MgCl2、dNTP和引 物的浓度分别为2.0、0.2 mmol·L-1和0.2 μmol·L-1,以 及0.5 UTaq DNA聚合酶和50ng DNA模板。最适反应 程序为先在94 ℃进行4 min 预变性;随后以94 ℃变 性45 s,53.7 ℃退火30 s,72 ℃延伸90 s为单个循环步 骤,循环扩增25 次;最后在72 ℃下延伸扩增10 min。 最佳反应体系和反应程序的PCR 效果见图 8。

|
| 图 8 最佳反应体系和反应程序的PCR效果 Figure 8 PCR result of the optimum PCR reaction system and procedure |
基于传统微生物分析方法的制约,Olsen 等[12]在 1986 年提出运用核糖体RNA 技术来研究微生物多 样性,从此进入了以分子生物学、生物标记等技术为 基础的现代微生物分析时代。在成分复杂的发酵床垫 料中含有大量的多糖、多酚、单宁、色素以及腐植酸, 在微生物DNA提取过程中,这些物质的存在会严重 影响DNA的质量和纯度,能否有效地去除这些物质 关系到整个DNA 提取过程的成败[2]。
在以微生物基因组DNA 为模板,扩增16S rDNA 的过程中,模板DNA 的质量是决定PCR 成败与否的 关键之一。若模板量太少,则引物与模板不能有效配 对,扩增效率低下;而若模板量太高,在退火过程中由 于模板密集导致引物难以找到结合位点,从而PCR 产量过低。引物是得到优质的特异性强PCR 产物的 关键。引物浓度偏高会发生错配和非特异性扩增,影 响靶序列的PCR 产量和质量,而且过多引物形成二 聚体造成资源浪费;浓度偏低会导致引物与模板结合 效率低,靶序列PCR产物量减少。Taq DNA聚合酶在 PCR 反应中起到非常关键的作用,但以适量为止,过 多添加Taq DNA 聚合酶不仅造成药品的浪费,而且 会产生非特异性扩增,导致靶序列PCR 产物产量和 质量的下降;低浓度Taq DNA 聚合酶会影响合成效 率,导致靶序列PCR 扩增产物减少。退火温度是PCR 反应过程中最关键的环节,受不同靶DNA 序列GC 含量的不同,导致它们PCR 扩增的退火温度差异较 大。通常GC 含量越高为了有效解链,需要越高的退 火温度。由于循环次数的增加会导致非特异性产物的 增加,所以确定其他反应参数之后进行优化是比较合 理的,以更好地提高靶序列PCR 产物的质量[4, 13]。 4 结论
为了更好地研究复杂多变的发酵床垫料中微 生物的多样性,本文对发酵床垫料中微生物总DNA 的提取方法进行了探索,同时建立与优化了微生物 16S rDNA PCR 反应条件,为揭示发酵床垫料中微生 物群落的多样性及其动态变化规律奠定了理论基础。
PCR 反应过程中各参数是相互依存的,退火时, 引物与模板DNA 结合,在Taq DNA 聚合酶和dNTP 存在下进行延伸,Mg2+是Taq 酶的激活剂。本文从 PCR 反应的稳定性和经济性因素的角度考虑,确定了 发酵床垫料中微生物16S rDNA PCR 扩增的最佳反应 体系和条件,25 μL 的最佳反应体系为:10×PCR buffer 2.5 μL、MgCl2 2.0 mmol·L-1、dNTP 0.2 mmol·L-1、 引物0.2 μmol·L-1、Taq DNA 聚合酶0.5 U、DNA 模板 50 ng;最佳反应程序为:在94 ℃下进行4 min预变性; 随后循环扩增25 个循环(包括94 ℃变性45 s,53.7 ℃ 退火30s,72℃延伸90 s);最后在72℃下延伸10 min。
| [1] | 王迪. 猪用生物发酵床垫料中微生物群落多样性变化及芽孢杆菌分离与鉴定[D]. 武汉: 华中农业大学, 2012: 12-13.WANG Di. The change of microbial community diversity in deep-littersystem and the isolation and identification of Bacillus[D]. Wuhan: Hua-zhong Agricultural University, 2012: 12-13.(in Chinese) |
| [2] | 刘云浩, 蓝江林, 刘波, 等. 微生物发酵床垫料微生物总 DNA 提取方法的研究[J]. 福建农业学报, 2011, 26 (2): 153-158.LIU Yun-hao, LAN Jiang-lin, LIU Bo, et al. Extraction methods forbeddingmicrobial DNAfromfermentation bed[J]. Fujian Journal of Agri-cultural Sciences, 2011, 26 (2): 153-158. (in Chinese) |
| [3] | 张邑帆, 卢茵, 黄微, 等. 发酵床垫料复合菌剂优化组合的研究[J]. 现代畜牧兽医, 2012 (2): 51-54.ZHANG Yi-fan, LU Yin, HUANG Wei, et al. Optimized combination ofdifferent microorganisms of fermentation mattress[J]. Modern Journal ofAnimal Husbandry and Veterinary Medicine, 2012 (2): 51-54. (in Chi-nese) |
| [4] | 邸宁, 刘志民, 马焕普. 研究土壤微生物多样性的 PCR 条件优化[J]. 安徽农业科学, 2011, 39 (23):14059 -14061, 14064.DI Ning, LIU Zhi-min, MA Huan-pu. Optimization of PCR condition insoil microbial diversity study[J]. Journal of Anhui Agricultural Sciences,2011, 39 (23): 14059-14061, 14064. (in Chinese) |
| [5] | 毕泗伟, 吴祖芳, 虞耀土. 16S rDNA 基因文库技术分析发酵床细菌群落的多样性[J]. 宁波大学学报: 理工版, 2013, 26 (1): 18-22.BI Si-wei, WU Zu-fang, YU Yao-tu. The bacteria diversity of fermen-tation bed in pig-farming by 16S rDNA gene clone library analysis[J].Journal of Ningbo University: NSEE, 2013, 26 (1): 18-22. (in Chinese) |
| [6] | Porteous L A, Armstrong J L. Recovery of bulk DNA from soil by a rapid,small-scale extraction method[J]. Curr Microbiol, 1991, 22: 345-348. |
| [7] | Ogram A, Sayler G S, Barkay T. The extraction and purification of micro-bial DNA from sediments[J]. Journal of Microbiological Methods, 1987, 7(2-3): 57-66. |
| [8] | Verma D, Satyanarayana T. An improved protocol for DNA extractionfrom alkaline soil and sediment samples for constructing metagenomiclibraries[J]. Appl Biochem Biotechnol, 2011, 165 (2): 454-464. |
| [9] | Yang Zh H, Xiao Y, Zeng G M, et al. Comparison of methods for totalcommunity DNA extraction and purification from compost[J]. Appl Mi-crobiol Biotechnol, 2007, 74 (4): 918-925. |
| [10] | 卢圣栋. 现代分子生物学实验技术 (第 2 版) [M]. 北京: 中国协和医科大学出版社, 1999: 458-463.LU Sheng-dong. Current protocols for molecular biology(2nd edition)[M]. Beijing: China Medical University Publishing House, 1999: 458-463. (in Chinese) |
| [11] | 郭凌飞, 邹明宏, 曾辉, 等. 澳洲坚果 ISSR-PCR 反应体系的建立与优化[J]. 林业科学, 2008, 44 (5): 160-164.GUO Ling-fei, ZOU Ming-hong, ZENG Hui, et al. Establishment andoptimizationof ISSR-PCRsystem in macadamia[J]. ScientiaSilvae Sini-cae, 2008, 44 (5): 160-164. (in Chinese) |
| [12] | Olsen G J, Lane D J, Giovannoni S J, et al. Microbial ecology and evolu-tion: a ribosomal RNA approach[J]. Ann Rev Microbiol, 1986, 40: 337-365. |
| [13] | 黄夕洋, 唐辉, 孔德鑫, 等. 广西甜茶 ISSR-PCR 反应条件的建立与优化[J]. 基因组学与应用生物学, 2013, 32 (4): 526-532.HUANG Xi-yang, TANG Hui, KONG De-xin, et al. Establishment andoptimization of ISSR-PCR reaction conditions in Rubus suavissimus S.Lee[J]. Genomics and Applied Biology, 2013, 32 (4): 526-532. (in Chi-nese) |